Analytical Laboratory Services
ICH Stability Studies
Stability Studies: GMP and non-GMP
ICH Stability Studies are a critical part of the drug development process. The results of these studies are used to determine the retest interval for an Active Pharmaceutical Ingredient (API) and dictate the shelf life for a drug product. Stability studies are typically initiated with material produced from either the first demonstration batch or from the batch used to support toxicology studies. The stabilities studies are then continued with each GMP batch. For cost-efficiency, an abbreviated study can also be performed when multiple batches are made from the same process.
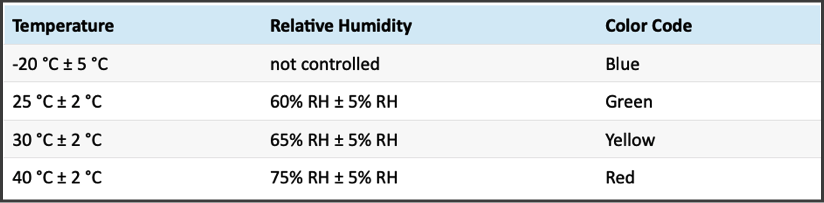
Stability Chambers Set to ICH Conditions
EZ Labs has multiple stability chambers set to ICH conditions for long-term and accelerated stability studies. Although typically conducted during forced degradation, photostability studies may also be considered as part of the stability study protocol.
Color-Coding for Accuracy
Color coding is used for both chamber and sample labeling, allowing chemists to easily recognize the relevant condition and reduce the potential for human error.
ICH Guideline Test Intervals
Test intervals typically follow ICH guidelines and may be modified as deemed appropriate for clinical APIs or as requested by the study sponsor. Backup samples are normally included for possible future investigation or if the study is extended.
Stability Study Quality Attributes
The quality attributes included in EZ Labs stability studies are those specifications (or other characteristics) of the API susceptible to change on storage. Our testing also includes a visual assessment of any interactions of the API with the container or closure mechanism.
A phase-appropriate study and protocol is provided by EZ Labs and subject to review by the client.
Stability Study Analysis & Reporting
In the event that results show little or no change (i.e. a clear trend with time), the retest interval or shelf-life is reported based in accordance with ICH guidelines.
If any test shows variability or an apparent trend with time, analysis may be used to establish a proposed retest interval that is consistent with the Q1E guidance.
Finally, stability study summary reports are provided to the client with the recommended storage conditions and relevant statistical analyses.

Method Development and Forced Degradation
Our Typical Method Development Projects
❖ Development of HPLC/UPLC assay and impurities methods supporting various stages of drug development programs
❖ Forced degradation studies and assessment of the degradation pathways of the NCE (including ID of the major degradation products)
❖ Development and qualification of LC/MS methods with SIM mode (or other methods as appropriate) for quantification of PGIs (potential genotoxic impurities) at the ppm levels
❖ Chiral HPLC method development and qualification
❖ Development of dissolution methods for various stages of drug development (including appropriate discriminating methods in support of NDA)
❖ GC methods supporting analysis of various volatile compounds and drug product residual solvents, following ICH guidelines
❖ Re-development, troubleshooting and fixing deficiencies of the existing methods. Conversion of long HPLC assay/impurities methods (sometimes with several actives) into shorter and more efficient UPLC methods
Forced Degradation and Understanding of the Degradation Pathway
Forced degradation is an integral part of impurity evaluation in method development, which is undertaken to facilitate development and to demonstrate the suitability of stability indicating methods. This is particularly important when little information is available about potential degradation products.
Forced degradation studies generally rely upon conditions that promote thermolytic, hydrolytic, oxidative, and photolytic degradation mechanisms. The goal of these studies is to generate a primary degradation profile that mimics or exceeds what would be observed in accelerated stability studies conducted under ICH conditions.
EZ Labs scientists design experiments to generate similar amounts of degradants to those occurring in stability studies. A team of analytical chemists can thus achieve a comprehensive understanding of the relevant degradation pathway.
Chiral Separations
The presence of stereocenters in intermediates and Active Pharmaceutical Ingredients (APIs) add an additional layer of required analytical understanding, monitoring and control. Each enantiomer or diastereomer may have great variations in efficacy and toxicity. In 1992 the Food & Drug Administration (FDA) issued a series of guidelines for the development of single enantiomers and racemates in pharmaceuticals.
We can rapidly screen a wide selection of chiral columns and then further optimize the resulting chiral HPLC method.
Our team of analytical chemists with extensive experience with poly-chiral compounds facilitates efficient and rational method development / optimization.
Method Development with HPLC-CAD
Charged Aerosol Detector (CAD) is a universal detector and (in conjunction with HPLC) can be used to measure all non-volatile (and some semi-volatile) compounds. This renders it extremely useful in the evaluation of purity and quantification of impurities for new drug candidates or intermediates lacking a significant UV chromophore.
EZ Labs successfully developed and qualified CAD based methods for a number of our clients and thus has a deep-rooted experience base in gaining invaluable knowledge with method development / optimization / troubleshooting with this mode of detection.
Formulation Development Studies
EZ Labs performs the following studies in support of all stages of formulation development process:
Excipient Compatibility
Study of drug-excipient compatibility is an important phase in the pre-formulation stage of drug development. The potential interactions between drugs and excipients have effects on the chemical, physical, bioavailability and stability of the dosage form. EZ Labs provides drug-excipient compatibility studies to provide data for drug-excipient interaction which can further help in selection of excipient for the development of stable dosage form.
Solubility Screening
Our Scientists perform solubility (including equilibrium pH solubility profiles) of lead discovery and development candidates and other IND-enabling studies.
Drug solubility is a fundamental property that has to be evaluated in the early stages of drug discovery. Various parameters will have an effect, such as the polarity of both the drug and the solvent, drug particle size, in addition to the parameters associated with the solution process, such as the temperature and agitation.
Early Development Stability
Early in development, pharmaceutical research organizations develop products with a primary focus on patient safety. Data are generated at appropriate storage conditions to demonstrate or support the stability of the drug substance and product to assure product quality through the clinical study period.
Photostability Studies
The intrinsic photostability characteristics of new drug substances and products should be evaluated to demonstrate that, as appropriate, light exposure does not result in unacceptable change. For confirmatory studies, samples should be exposed to light providing an overall illumination of not less than 1.2 million lux hours and an integrated near ultraviolet energy of not less than 200 watt hours/square meter (refer to ICH Q1B guidelines).
Thermal Cycling and Freeze-Thaw Studies
Temperature sensitive medicinal products should be transported in a manner which ensures product quality is not adversely affected. Transportation of product typically takes place within a commercial environment. Factors such as traffic events and unforeseen weather conditions can delay shipments and have a deleterious effect on the product. The range encountered during transportation may differ from that which is specified for long term storage, ascertained from stability studies. To understand the effect these excursions may have on a product, Thermal Cycling studies are completed. Also known as freeze thaw studies, they are used to evaluate the potential effects of temperature deviations that may occur during the transport process.
Reference Standard Certification
What are Reference Standards?
A Reference standard is defined as a highly characterized material suitable to test the identity, strength, quality and purity of substances for pharmaceutical use and medicinal products.
At EZ Labs, reference standards characterized via a suite of analytical and NMR techniques. A characterization report is written based on the filing requirements (upon request from the client).
EZ Labs scientists divide reference standards into two groups: Retention Markers and Quantitative Reference Standard.
Retention Time Markers
These are typically starting materials, impurities, intermediates, and drug substances used qualitatively in test methods to mark retention times and identify peaks, as components of mixtures to establish resolution, or to establish identity via spectral comparison. They are not used for quantitative weight % assays.
Quantitative Reference Standards
These reference standards are typically starting materials, intermediates, or drug substances used in test methods as working standards for quantitative weight % assay determinations of major components.
• Quantitative reference standards can also be starting materials, intermediates, or other impurities used in test methods as working
standards for weight % impurity determinations.
• Quantitative reference standards can also serve as retention markers and similar qualitative reference standards.
Characterization of Reference Standards
The following tests are typically used to characterize reference standards intended for use in qualitative tests. This list may be altered as judged appropriate for a given project or as requested by a client:
• Physical appearance
• Two identification tests, typically including NMR, IR, or Mass spectrum
• Chiral Purity
The following tests are typically used to characterize reference standards intended for use in quantitative weight % assays for both, major components and impurities. This list may be altered as judged appropriate for a given project or as requested by a client:
• Physical appearance
• Two identification tests, typically including NMR, IR, or Mass spectrum
• Area % purity by HPLC or GC, with impurity profile
• Chiral Purity
• Residual solvents by GC
• Water content by Karl-Fischer titration
• Melting point (for solids)
• Inorganic impurities (residue on ignition / sulfated ash)
• Solid form by X-ray diffraction (for solids)
• Assigned purity or purity factor
In all cases, a Certificate of Analysis (COA) will be provided to the client.
GMP Analytical Services
EZ Labs LLC recently relocated to a larger space, with an increased footprint of 8,000 sq. ft., that added QC laboratories, stability storage, document control and IT areas.
In addition, a phase-appropriate quality management system (QMS) was implemented to expand its capacity and support early phase GMP activities. The QMS includes a formal equipment qualification and preventative maintenance program, reference standard qualification and management program, sample management, and a controlled documentation and a comprehensive training program, among others.
The existing QMS enables EZ Labs LLC to develop and execute phase-appropriate method validations, transfers, and support GMP release and stability activities throughout the development lifecycle, including Phase 1-2 clinical trials.
Method Validation & Verification
Method Validation & Verification are experimental processes of establishing that the performance characteristics of a method meet the requirements of the intended analytical application.
Methods can be developed in-house, adopted from the client, or transferred to a client’s designated laboratories. This work is performed as needed and in a phase-appropriate fashion prior to tox batch delivery. Method verification / validation occurs prior to the subsequent GMP delivery.
Method verification / validation protocols are prepared following a phase-appropriate strategy informed by the EZ Labs in-house SOP, written according to ICH Q2 guidance and current USP Chapters <1225> and <1226>. These protocols are available to be reviewed by the client.
Method Validation
Method validation applies to newly developed analytical methods. Typical analytical characteristics evaluated during method validation may include:
• Accuracy
• Precision
• Specificity
• Detection Limit
• Quantitation Limit
• Linearity
• Range
Robustness may also be determined during method development. Solution stability will be established to ensure that analysis can be conducted within a given time frame without stability issues.
Method Verification
Method verification applies to analytical methods described in USP-NF, EP or validated methods transferred from other labs. Method verification consists of assessing selected analytical characteristics to generate appropriate relevant data (as opposed to repeating the entire validation process).
Method Transfer
Method Transfer applies to analytical methods transferred between external laboratories and EZ Labs. This ensures that the receiving laboratory has the procedure, knowledge and ability to perform the transferred analytical procedure as intended. Method transfer may be performed and demonstrated in various ways, including:
• comparative testing
• co-validation between two or more laboratories
• complete or partial method validation or revalidation
• the omission of formal transfer, sometimes termed the transfer waiver.
The approach proposed by EZ Labs will be subject to approval by the client.
Impurity Isolation and Structure Elucidation
Isolation Strategies
In terms of isolation strategies, we force degrade the main compound to determine if the impurity is increased. This degraded material with increased amount of impurity will make the isolation by LC-UV-MS preparatory chromatography much easier. We can also analyze any existing mother liquor’s that may be enriched in desired impurities and use the mother liquors for impurity isolation if they contain larger amounts of desired impurities. These techniques have been employed in recent projects for isolating impurities that are at the 0.1 % level in the main compound.
LC-MS, NMR and LC-HR/MS Data Acquisition & Interpretation
Once a suitable amount of a relatively pure isolate (greater than roughly 85%) is obtained, 1D/2D NMR data acquired using Bruker DRX-500 500 MHz NMR spectrometer. The data sets not only encompass conventional proton-proton and proton-carbon correlation, but also proton-nitrogen correlation experiments to allow for unambiguous structural assignments. The instrument is equipped with a 3 mm inverse probe that can be used for samples that are on the sub-milligram level.
Derivation of the Structural Formula
Once a complete NMR data set is obtained, a structural formula can be determined that fits the NMR data, as well as the mass spectrometry parent and fragment structures. Also, high resolution MS and fragmentation MS data can be used, if needed. Ultra-High Resolution Bruker Impact II QTOF LC/MS provided through our partners is the latest innovation in Bruker’s unique UHR-QqTOF mass spectrometry product with industry-leading > 50,000 Full-Sensitivity Resolution (FSR). It opens up enhanced analytical performance levels for all applications where trace analysis from complex, high-background matrices is a challenge. A good structural formula would be consistent with all the NMR and mass spectrometry data. As an add-on bonus, we can recover the impurity from solution and provide it as an HPLC marker for analytical development.